










购物车
- 全部删除
 您的购物车当前为空
您的购物车当前为空

我们很想知道您的意见反馈,所以我们在每个页面上都梳理出一个反馈按钮。
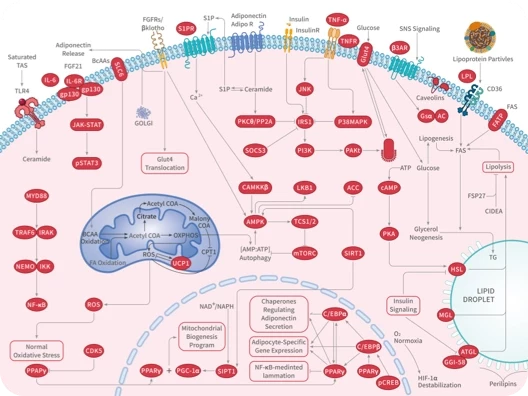
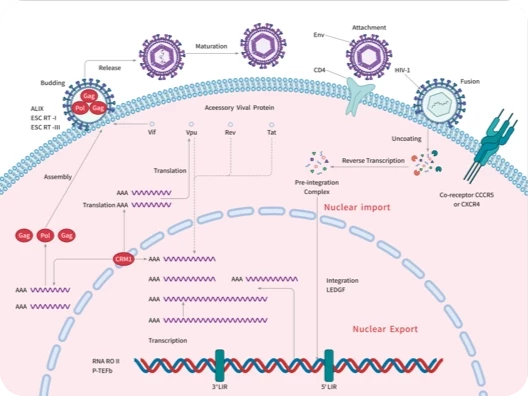
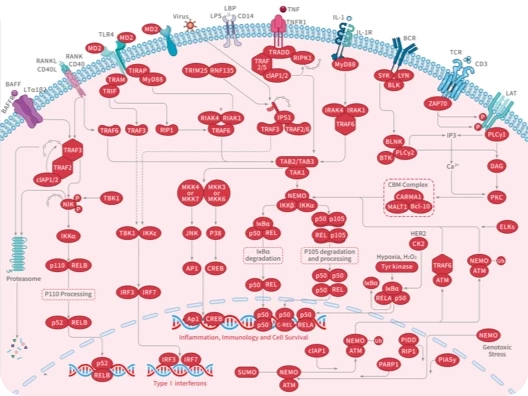
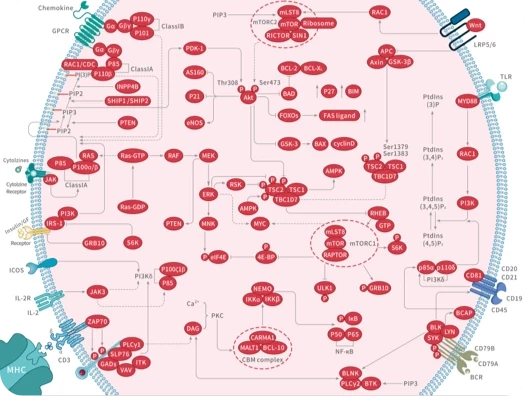
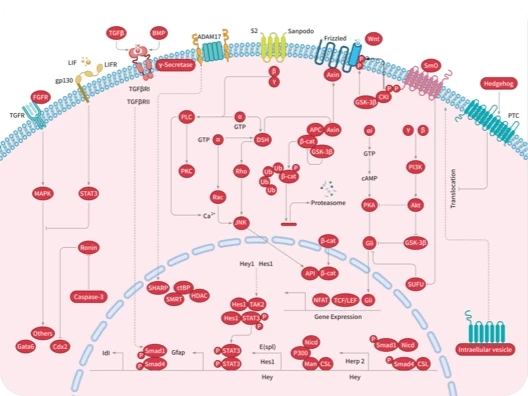
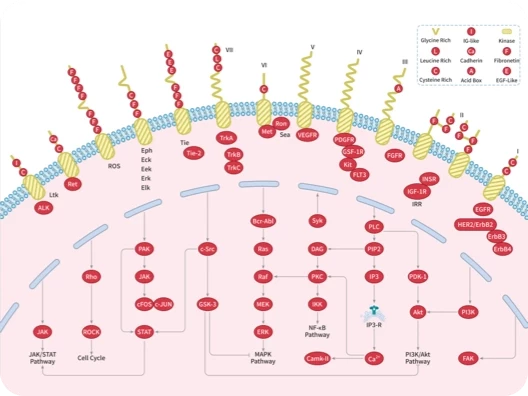
在新一期的 Signal Transduction and Targeted Therapy 杂志中,四川大学华西医院联合成都中医药大学的研究团队介绍了多种“不可成药”蛋白的药物发现策略,并总结了已成功开发出的多种治疗方法。这项研究有望进一步激发药物设计和应用领域的思考。

随着对“不可成药“蛋白研究的不断深入,具有不可成药特征的分子可分为以下几类:
1)小GTP酶(Small GTPases)
2)磷酸酶(Phosphatases)
3)转录因子(Transcription factors,TFs)
4)表观遗传靶点(Epigenetic targets)
5)其他蛋白
根据不可成药蛋白的各种特性,形成了一些主要的药物设计策略,包括 共价调控、变构抑制、PPI 抑制、靶向蛋白调控、基于核酸的方法、免疫治疗等,快和小陶一起来看看吧~
共价抑制剂也叫做不可逆抑制剂,是一类通过温和反应性官能团形成的共价键与靶蛋白的氨基酸残基结合,以赋予额外亲和力的抑制剂。
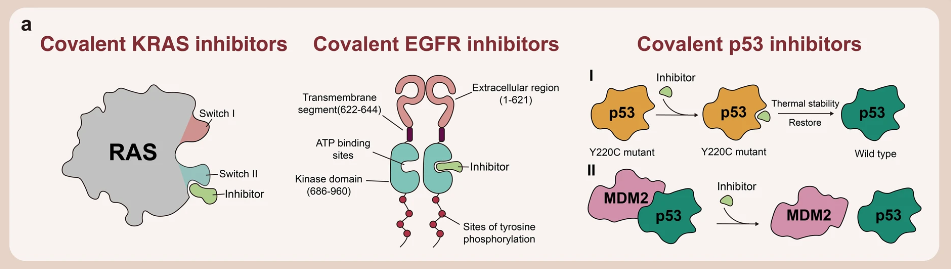
▲共价调节剂的结合模式
§
KRAS 共价抑制剂
KRAS 基因的全名叫 Kirsten ratsarcoma viral oncogene homolog,KARS 基因编码的蛋白是一种小 GTP 酶(smallGTPase),它属于 RAS 超蛋白家族。因为 KRAS 蛋白是一个没有明显结合位点的近球形结构,很难开发有效靶向和抑制其活性的化合物。然而,通过采用共价调控的方法,可以克服这一难题。
KRAS 共价抑制剂与 KRAS G12C 突变体的胱氨酸结合,可降低 GTP 和 KRAS 之间的亲和力,从而将 KRAS G12C 突变体锁定在失活状态。
目前已上市的共价 KRAS 抑制剂有 Sotorasib 和 Adagrasib。除此之外,还有 10 种 RAS 抑制剂正在临床中(如下表),28 种正在临床前开发中,详情点击“阅读原文“查看哦~
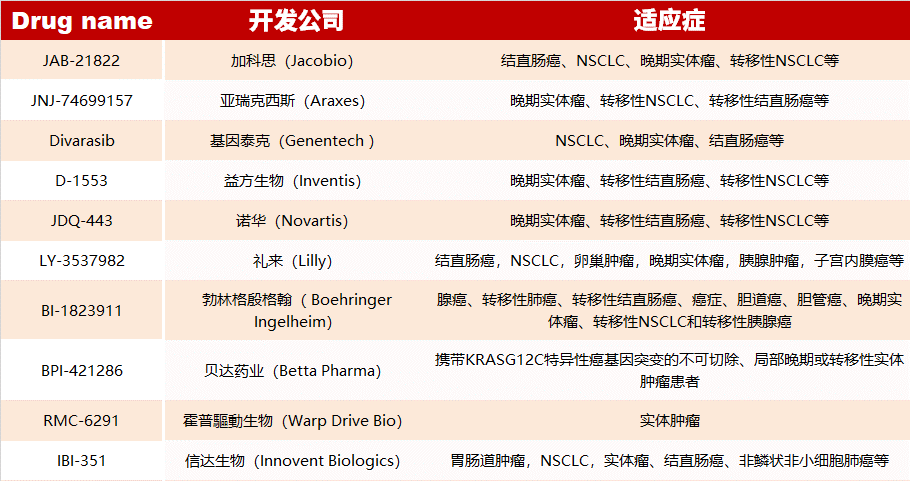
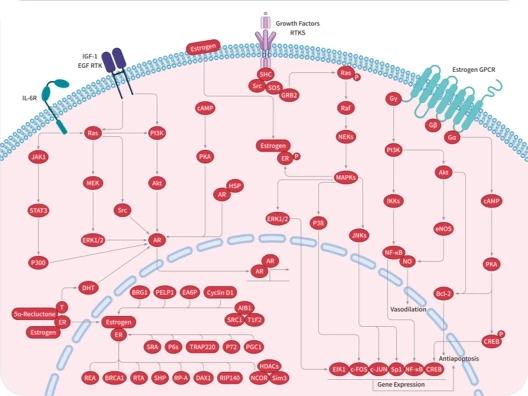
表皮生长因子受体(EGFR)是一种跨膜糖蛋白,存在于人体各组织细胞膜上,是受体酪氨酸激酶中的表皮生长因子受体(HER)家族中的一员,在调节细胞增殖、存活和分化过程中起着重要作用。
EGFR 的靶向药物是酪氨酸激酶抑制剂(TKI),其抑制细胞中的激酶,从而阻止它们激活 EGFR 信号通路。第一代 EGFR TKI,如 gefitinib 和 erlotinib,以非共价结合的方式选择性地与 EGFR 酪氨酸激酶的 ATP 结合位点结合,从而抑制 EGFR 磷酸化,显著延缓临床 NSCLC 靶向治疗的疾病进展。但在使用第一代 EGFR TKIs 后,会出现耐药性,导致肿瘤再次进展。因此有了第二代、第三代 EGFR 共价抑制剂的出现。
目前已上市的 EGFR 共价抑制剂如下:

除此之外,还有一些 EGFR 共价抑制剂正在申请批准中,例如 avitinib, oritinib, sunvozertinib 和 rezivertinib。一些 EGFR 共价抑制剂正在进行临床试验:如Olafertinib (CK-101/RX518) 、Nazartinib (EGF816, NVS-816) 、Allitinib (AST-1306)。
P53 蛋白是一种转录因子和肿瘤抑制蛋白,可结合 DNA 并调控多种转录靶标,响应细胞应激或 DNA 损伤,协调细胞周期阻滞、DNA 修复、代谢改变、凋亡等过程,防止肿瘤形成。大约一半的人类癌症中都存在 P53 基因的突变。
癌症中 TP53 突变的主要后果是肿瘤抑制功能的丧失,需要治疗性地重新激活该蛋白,而大多数与癌症有关的小分子药物实际上是通过抑制蛋白的过度活性来发挥作用。因此,P53 长期以来一直被认为是“不可成药”的。
2022 年,Kevan M. Shokat 的团队开发了一种小分子共价抑制剂 KG13。KG13 专门设计用于与 P53-Y220C 突变体结合,可将 P53 靶基因激活,抑制肿瘤细胞生长和增加半胱天冬酶活性。除了直接靶向 P53 外,还可以通过靶向 P53 的调节因子来达到治疗的效果。例如靶向 MDM2 的 NPD6878 (apomorphine)、和靶向 HDM2的 Compound 76。
当然,除了 KRAS、EGFR、P53 外,还有靶向其它“尚未成药“靶标蛋白的共价抑制剂正在开发中,如靶向 Myeloid cell leukemia-1 (Mcl-1) 的共价抑制剂 MAIM1、一种基于三价砷共价诱导剂开发的 PKM2 抑制剂 Compound 78 等等。
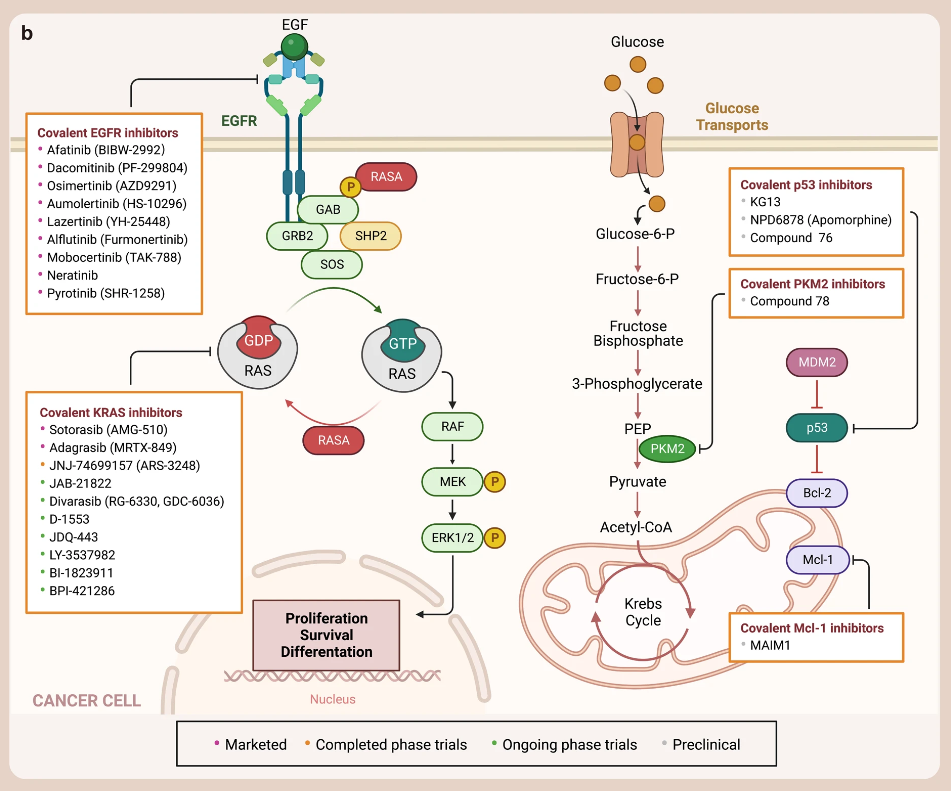
▲已上市、临床和临床前共价抑制剂的图谱
变构调节是一种直接有效的调控生物大分子功能的方法,通过变构改变生物大分子四级结构进而改变其生物功能,存在于各类细胞活动中。例如信号转导、酶催化、细胞代谢、基因调控等。变构调节剂也有着使”不可成药“靶点转化为”可成药“靶点的能力。
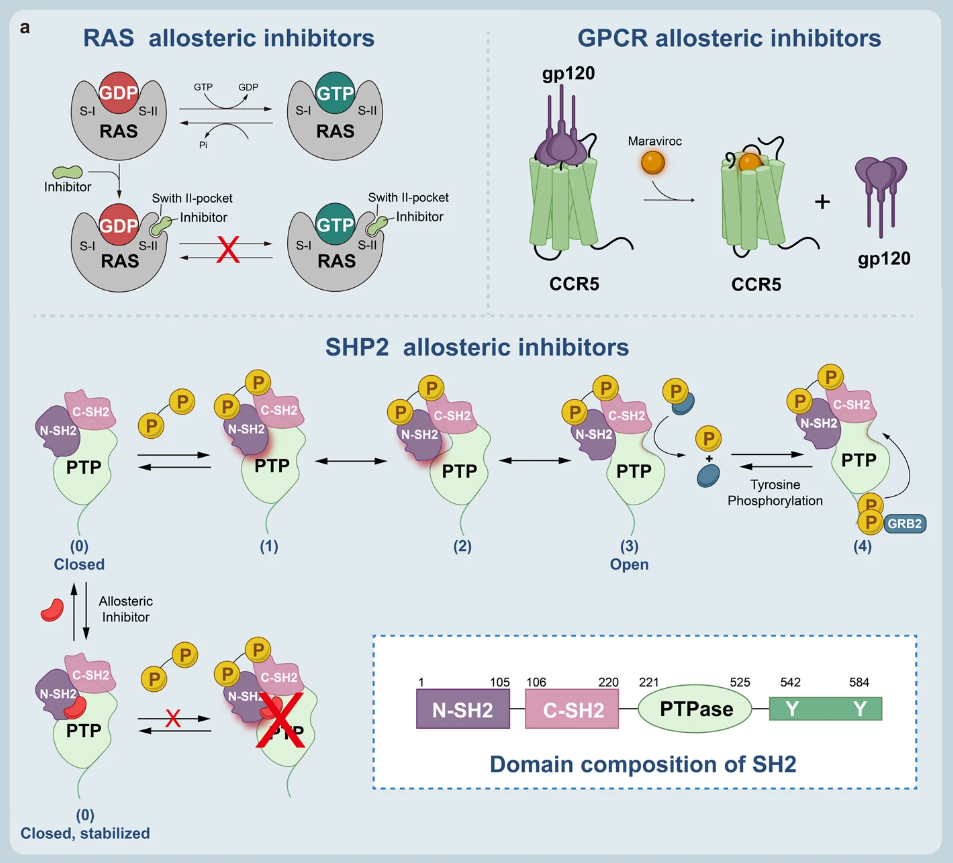
▲变构调节剂的结合模式
变构调节剂可以分为三类:
1)正变构调节剂(PAMs):可与受体结合改变受体对刺激的反应;
2)负变构调节剂(NAMs):通过降低配体的亲和力和/或功效来减弱信号传导的新型间接方法;
3)沉默变构调节剂(SAMs):通过改变蛋白质构象或功能来抑制目标蛋白质活性的药物或化合物。
通过应用变构调节的策略已开发了许多靶向 KRAS、SHP2 的药物,部分如下(更多信息可查看原文):
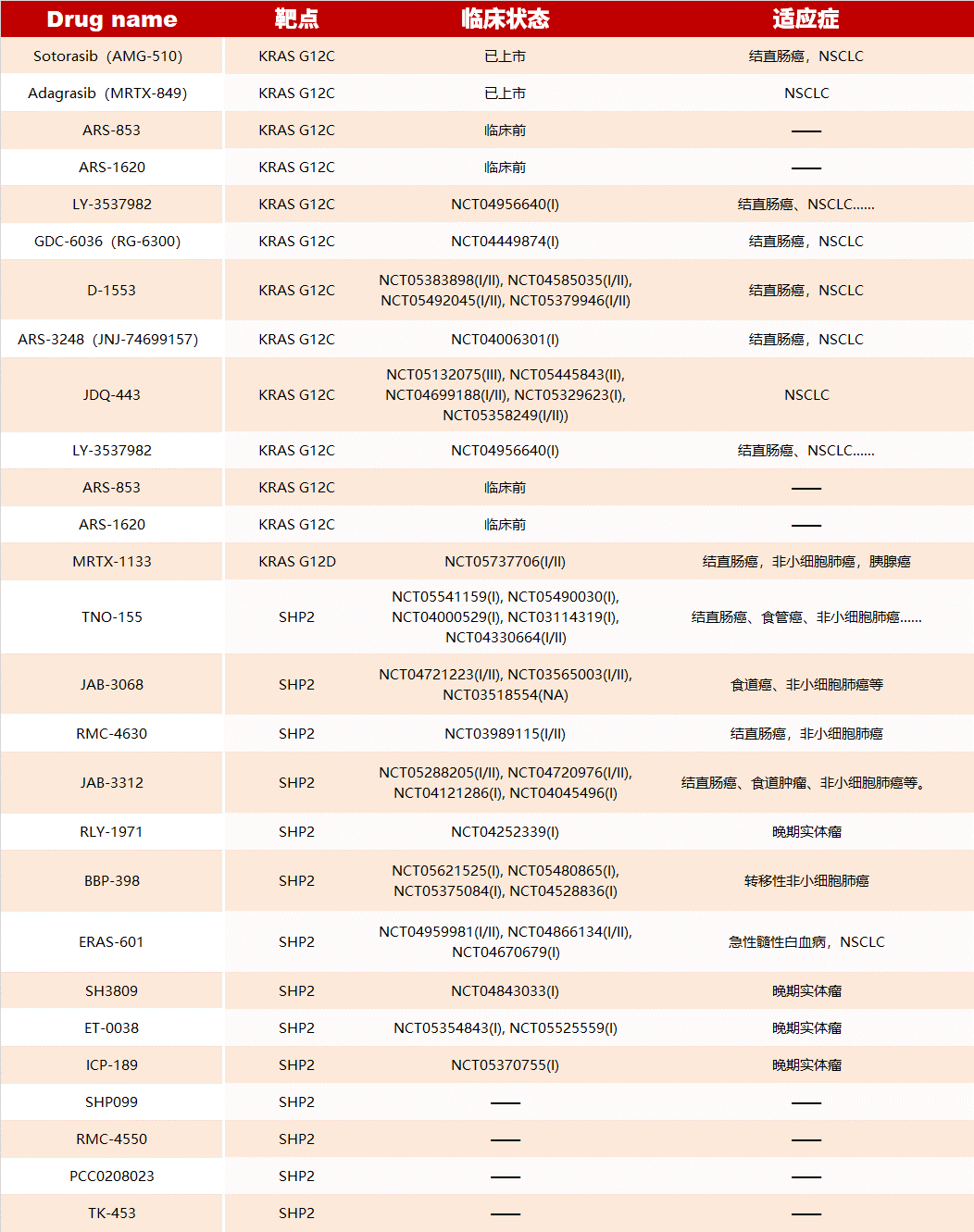
此外,已有一些 GPCR 变构调节剂获批上市,如 avacopan、cinacalcet、ticagrelor 和 maraviroc,还有数十种药物分子正在进行Ⅰ-Ⅲ期临床试验,数百种有潜力的 GPCR 变构调节剂正在进行临床前研究。
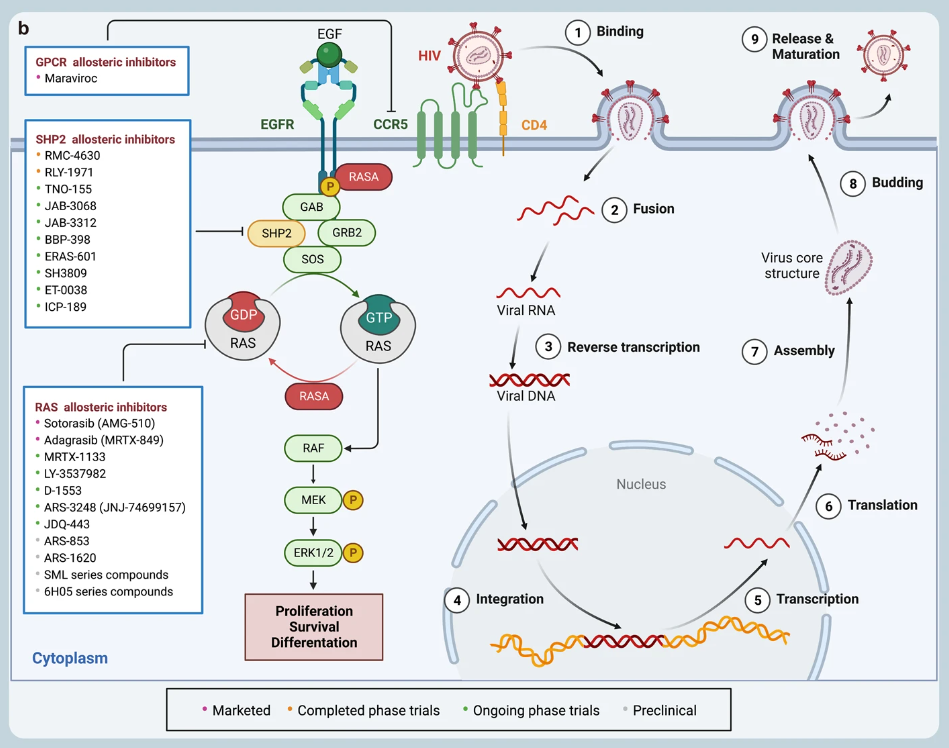
▲已上市、临床和临床前变构抑制剂的图谱
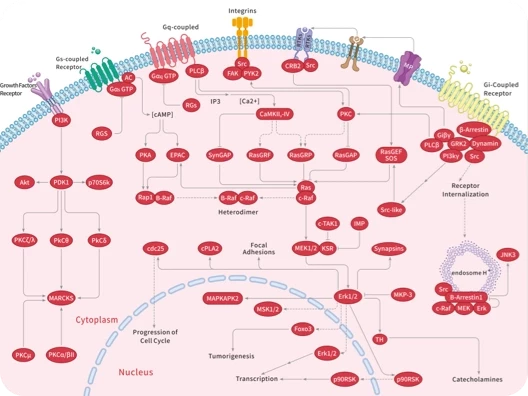
蛋白-蛋白相互作用(PPI)在众多生物过程中扮演着至关重要的角色,包括信号传导、细胞增殖、生长、分化和凋亡等。设计药物来干预 PPI 已经被证明是一种有效的策略,可以有针对性地作用于那些“难以靶向”的蛋白质。现有的PPI抑制剂可以分为小分子、抗体、肽和重组蛋白,每种类型都有其优点和缺点。

▲PPI调节剂的结合模式和机制
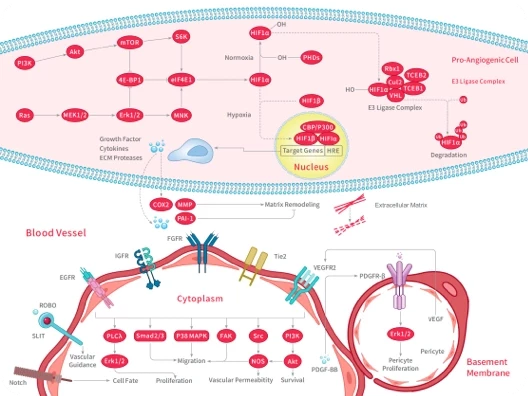
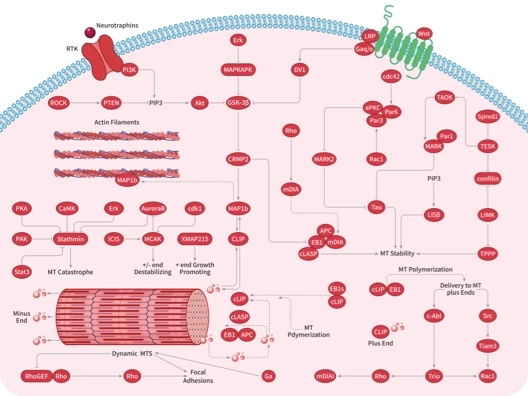
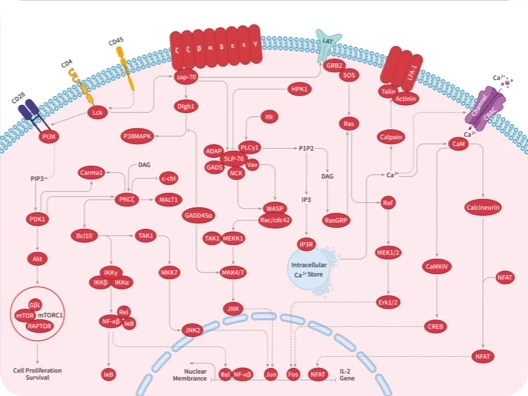
RAS 可通过结合 GTP 或 GDP 在活动状态和非活动状态之间切换,传递细胞生长和分化的信号。活化的 RAS 蛋白可以与许多下游效应蛋白相互作用,如 RAF 激酶、鸟苷核苷交换因子 SOS、磷脂酰肌醇 3 激酶(PI3K)和 Ral 鸟苷核苷酸解离促进因子(RalGDS)等。
目前应用 PPI 策略设计 RAS PPI 分子或药物有:

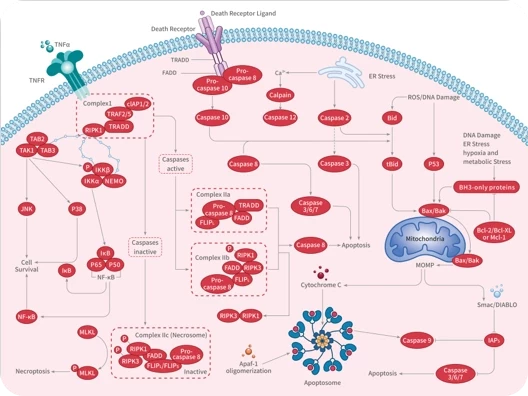
Bcl-2 家族在通过调控线粒体/细胞色素 C 介导的内源性凋亡信号转导途径中发挥关键作用,在各种血液恶性肿瘤中高度表达,包括慢性淋巴细胞白血病(CLL)和滤泡中心淋巴瘤(MCL)等。Bax 和 Bad 等促凋亡蛋白对细胞凋亡过程至关重要,但当它们与 Bcl-2 等抗凋亡蛋白结合时,它们的功能会被抑制,因此,阻止凋亡和抗凋亡蛋白之间的相互作用可以防止肿瘤细胞逃脱凋亡。
目前已开发了众多 Bcl-Bax 抑制剂,包括 ABT-199 (venetoclax),ABT-737, ABT-263 (navitoclax) 等。
MDM2 是 P53 最重要的负调节因子,可结合到 P53 的 N-末端的转录激活结构域,抑制 P53 的转录活性,抑制细胞生长,调控细胞周期并诱导细胞凋亡。因此,破坏 MDM2-P53 相互作用是治疗癌症的潜在途径。
目前,一系列 MDM2-P53 抑制剂已进入临床试验:
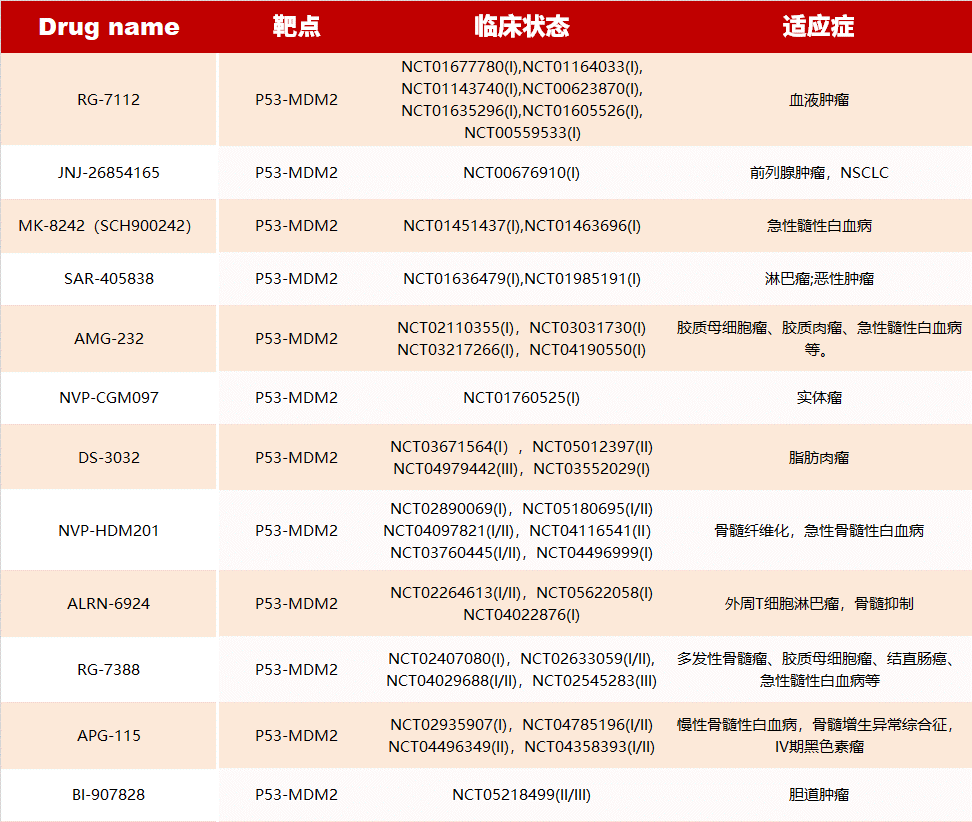
Myc 癌基因,包括其三个亚型 C-Myc、N-Myc 和 L-Myc,被认为是一种可以在不同类型的癌症中触发各种致癌转录程序的转录“放大器”。由于 Myc 缺乏结合小分子化合物的疏水口袋或凹槽,因此它也被认为是一种难以药物干预的靶点。迄今为止,尚未有获批上市的 Myc 药物,但有许多分子正在临床前研究中。

当然,以上仅是部分采用 PPI 策略针对“不可成药“蛋白的例子,更多的还有 YAP/TAZ、β-catenin、XIAP 等,小陶在这路就不一一叙述了,大家可以点击文末”阅读原文“查看详情~
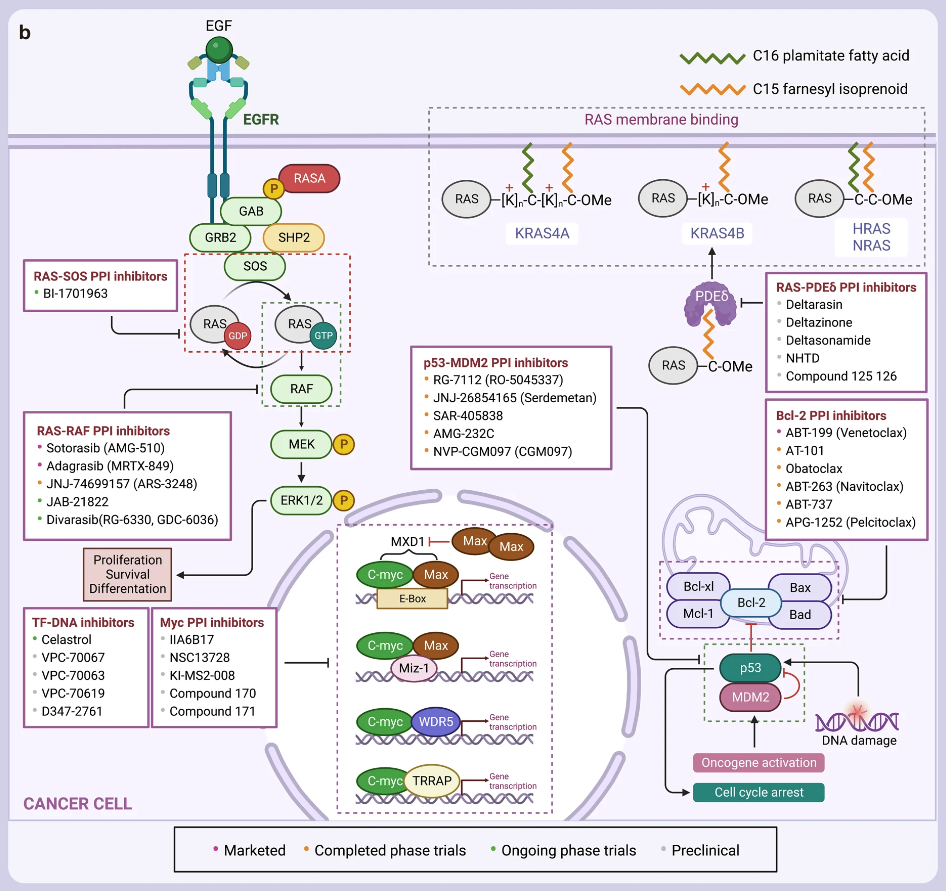
▲已上市、临床和临床前PPI抑制剂图谱
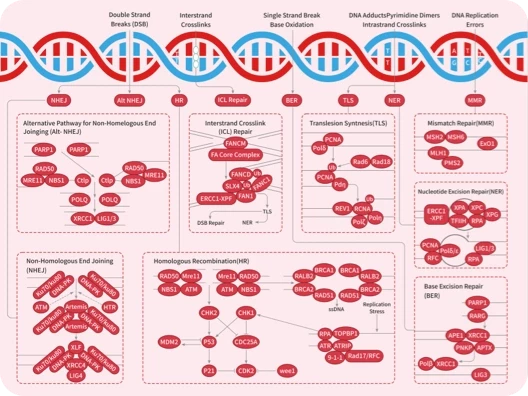
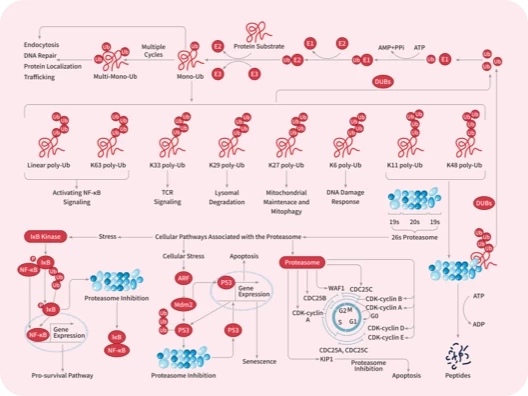
鉴于那些“不可成药”的蛋白质大多具有相似的特点,包括 表面平坦、缺乏活性位点 等问题,再加上蛋白质与疾病之间的直接关联,因此,通过直接调节与疾病相关的蛋白质已经被证明是一种非常有前景的治疗策略。根据疾病的机制,可以分为两个主要类别:蛋白质降解(TPD)和蛋白质稳定(TPS)。
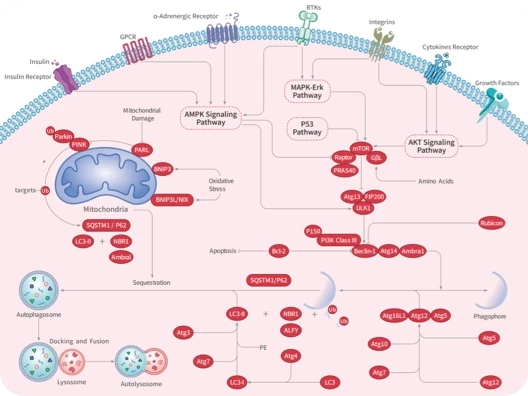
在这一领域表现较为出色的是蛋白降解靶向嵌合体(PROTAC)。PROTAC 是一种独创性的蛋白质调控方法,其核心作用机制是通过 泛素-蛋白酶体系统 (UPS) 同时靶向感兴趣的蛋白质和 E3 泛素连接酶,形成三元复合物,在这个过程中,目标蛋白质被泛素标记,促使其被蛋白酶体识别并降解。
PROTAC 技术也是靶向“不可成药“蛋白的有前途的方法。目前已有一系列的研究正在进行中:
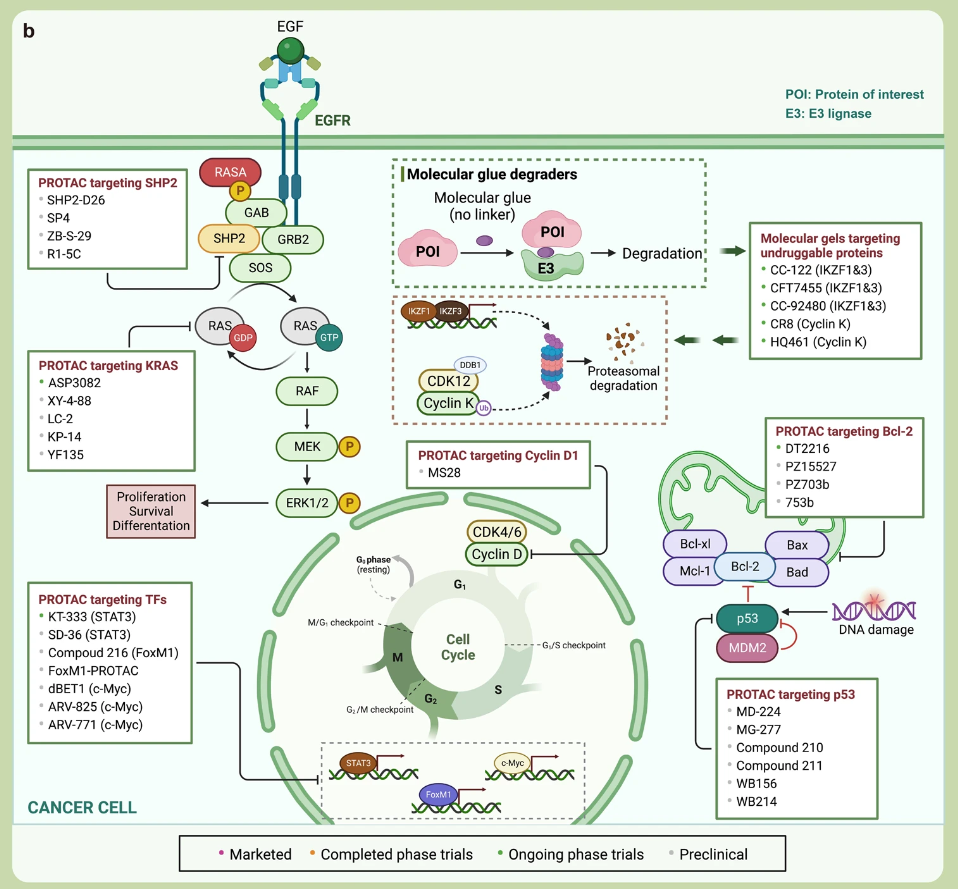
▲已上市、临床和临床前靶向蛋白质调控分子的图谱
除了 PROTAC 外,分子胶也是备受关注的靶向蛋白降解剂之一。在靶向蛋白降解 (TPD) 领域,分子胶通常是一种单价小分子 (<500Da),能通过重塑 E3 连接酶受体的表面促进目标蛋白(Protein of interest)的募集,进而推动目标蛋白的泛素化,随后被蛋白酶体降解。(关于 PROTAC 与分子胶的区别可知乎搜索以下文章查看哦)
《什么是分子胶,和PROTAC有什么区别?靶向蛋白降解剂的原理及开发?》
目前已有多种分子胶药物正在临床试验中:
PROTAC 的潜力并不仅限于此,科研人员已开发出了可以靶向“不可成药”蛋白的类 PROTAC 技术的变体。例如 P-PROTAC、PROTAB、CHAMP 以及基于溶酶体的 TPD 等技术。
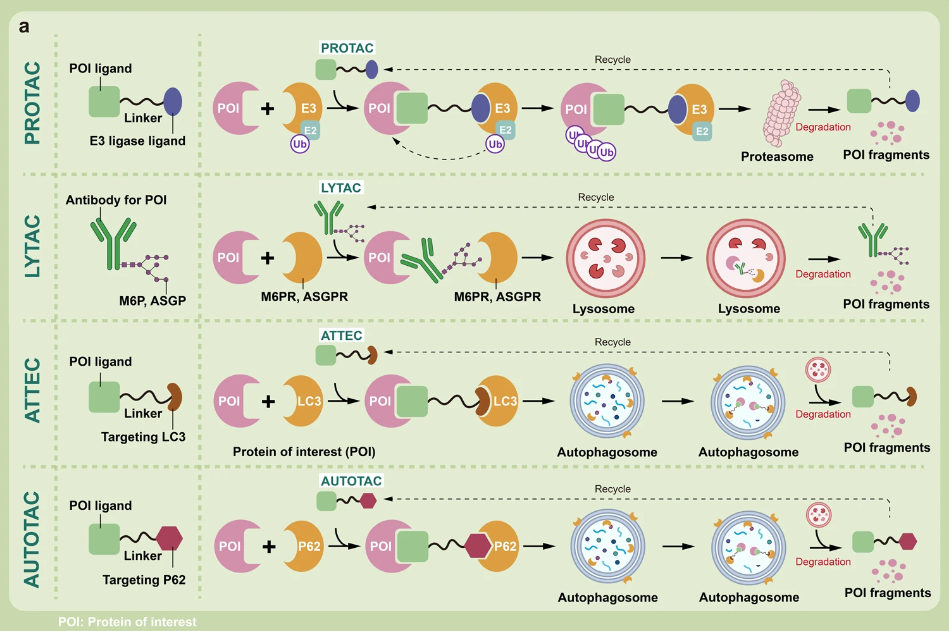
▲靶向蛋白质调控策略
尽管已经证明在某些疾病治疗中 TPD(蛋白质降解)是有效的,但蛋白质的过度降解可能会引发其他不良问题。因此,需要研发靶向蛋白质稳定的药物,以 抵消蛋白质的过度降解。研究人员正朝着这个方向不断努力。
其中比较有应用前景的是 去泛素酶靶向嵌合物(DUBTAC)。加州大学伯克利分校的 Nomura 团队受到 PROTAC 的启发,开发了 DUBTAC 用于靶向蛋白稳定(TPS),并创造了分子 NJH-2-057,这种 DUBTAC 可有效抑制 ΔF508-CFTR 的降解,提高蛋白质水平,并在囊性纤维化供体细胞中表现出很好的稳定性。

在此基础上,哈佛医学院的 Wenyi Wei 团队又开发了一种 TF-DUBTAC 技术,能够选择性地稳定致癌蛋白,包括 FOXO3A、p53 和 IRF3 等,是一种很有前景的治疗方法。
更多的靶向“不可成药”蛋白的方法,如基于核酸的疗法、免疫疗法、针对上下游效应物/辅因子的疗法等小陶就不一一讲啦,感兴趣的朋友可以点击“原文链接”查看原文哦~
总结
在药物设计策略方面,针对那些难以成药的蛋白质,目标和思路都至关重要。这些蛋白质与其他分子在生物网络中存在复杂的相互关系,因此,针对多种生物过程,如 蛋白质相互作用 和 RNA 介导的蛋白质表达,可能是一种有效的途径。此外,针对难以药物化的蛋白质本身,有针对性的 共价抑制剂、变构抑制剂 和 PROTAC 等新兴技术,可以通过控制或调节靶标蛋白的活性,甚至直接降解这些蛋白质,从而实现“不可成药”蛋白质的药物化。
TargetMol拥有 800+化合物库,其中有一些是致力于不可成药靶点的开发的,例如 PPI抑制剂库、KRAS靶向化合物库、表观遗传库、转录因子库 等,欢迎私信咨询。
参考资料:
[1] Xie, X., Yu, T., Li, X. et al. Recent advances in targeting the “undruggable” proteins: from drug discovery to clinical trials. Sig Transduct Target Ther 8, 335 (2023). https://doi.org/10.1038/s41392-023-01589-z
[2] Henning, N.J., Boike, L., Spradlin, J.N. et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat Chem Biol 18, 412–421 (2022). https://doi.org/10.1038/s41589-022-00971-2
[3] Jing Liu, Xufen Yu, He Chen, H. Ümit Kaniskan, Ling Xie, Xian Chen, Jian Jin, and Wenyi Wei
Journal of the American Chemical Society 2022 144 (28), 12934-12941. DOI: 10.1021/jacs.2c04824


科学新闻、观点和分析的重要汇总,每个工作日都会发送到您的收件箱.
 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询