我们很想知道您的意见反馈,所以我们在每个页面上都梳理出一个反馈按钮。
抑制剂 Cocktails
磁珠产品
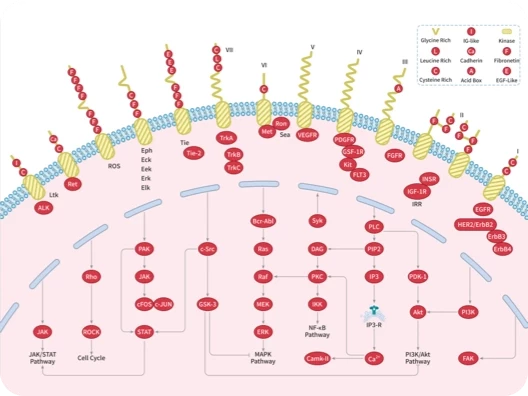
ALK
Bcr-Abl
BTK
EGFR
FAK
FGFR
FLT
HER
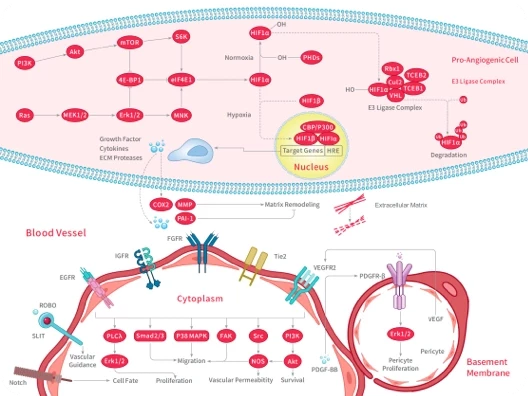
HIF
JAK
PDGFR
Src
Syk
VDA
VEGFR
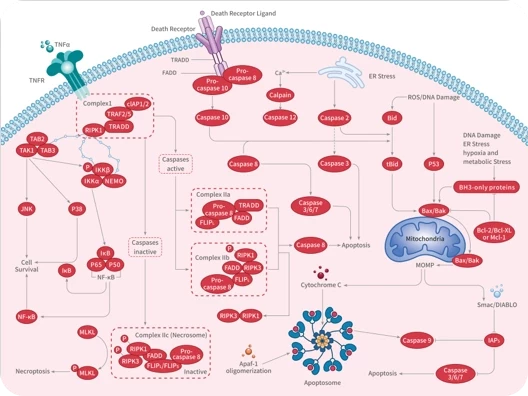
Apoptosis
ASK
BCL
Caspase
c-RET
DAPK
Fas/FasL
Ferroptosis
FKBP
FOXO1
IAP
Mdm2
Necroptosis
OXPHOS
p53
PD-1/PD-L1
PERK
Pyroptosis
RIP kinase
Survivin
TNF
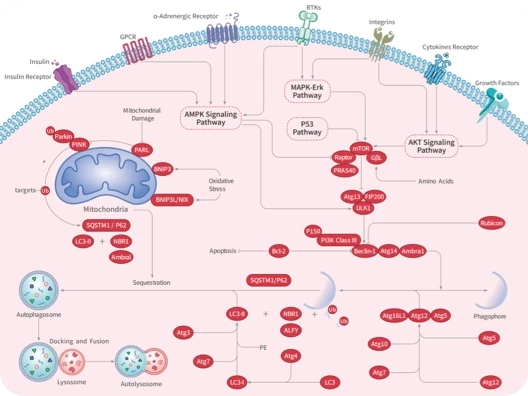
Autophagy
CXCR
LRRK2
lysosomal autophagy
Mitophagy
Antifolate
APC
Aurora Kinase
BMI-1
CDK
Cell Cycle Arrest
Chk
c-Myc
DHFR
DNA/RNA Synthesis
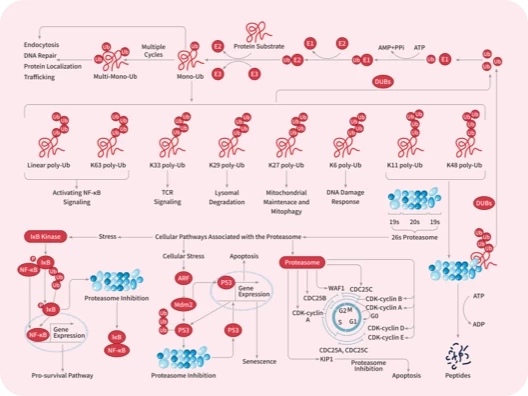
DUB
DYRK
Haspin Kinase
IRE1
KSP
LIM Kinase
Nucleoside Antimetabolite/Analog
PLK
Rho
ROCK
Serine/threonin kinase
Wee1
AMPK
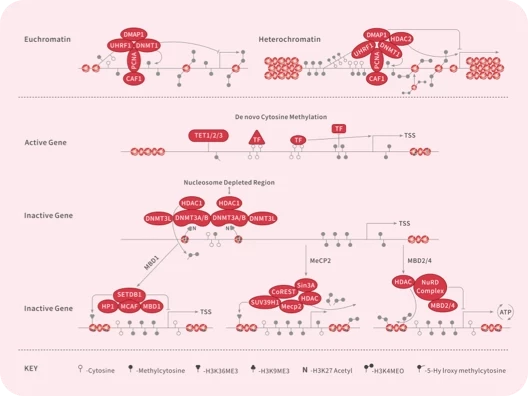
DNA Methyltransferase
Epigenetic Reader Domain
HDAC
HIF/HIF Prolyl-Hydroxylase
Histone Acetyltransferase
Histone Demethylase
Histone Methyltransferase
HuR
Methionine Adenosyltransferase (MAT)
PAD
PARP
Pim
PKC
Sirtuin
Akt
Arp2/3 Complex
Dynamin
Gap Junction Protein
HSP
Integrin
Kinesin
Microtubule Associated
Myosin
PAK
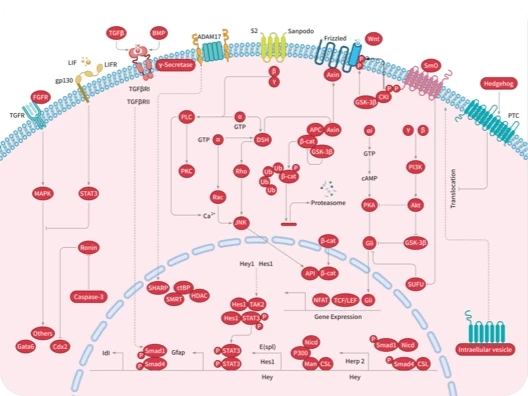
Wnt/beta-catenin
ATM/ATR
CRISPR/Cas9
DNA
DNA Alkylation
DNA Alkylator/Crosslinker
DNA gyrase
DNA-PK
MTH1
PPAR
Telomerase
Topoisomerase
Androgen Receptor
Aromatase
Estrogen Receptor/ERR
Estrogen/progestogen Receptor
Glucocorticoid Receptor
GPR
LHRH
Opioid Receptor
Oxytocin Receptor
RAAS
Reductase
Thyroid hormone receptor(THR)
5-HT Receptor
Adenosine Receptor
Adiponectin receptor
Adrenergic Receptor
Amylin Receptor
Annexin A
Apelin receptor
Arrestin
Bombesin Receptor
Bradykinin Receptor
cAMP
Cannabinoid Receptor
CaSR
CGRP Receptor
cholecystokinin
CRFR
Dopamine Receptor
EBI2/GPR183
Endothelin Receptor
GHSR
Glucagon Receptor
GNRH Receptor
GPCR19
GRK
GTPase
Guanylate cyclase
Hedgehog/Smoothened
Histamine Receptor
Kisspeptin
Leukotriene Receptor
LPA Receptor
LPL Receptor
Melanin-concentrating Hormone Receptor (MCHR)
Melanocortin Receptor
Melatonin Receptor
Motilin Receptor
Neurokinin receptor
Neuropeptide W
More...
AhR
AIM2
Antiviral
Arginase
Aryl Hydrocarbon Receptor
CCR
CD38
CD73
Cell wall
cGAS
Complement System
COX
Cytokine release
FLAP
Galectin
gp120/CD4
IFNAR
IL Receptor
Immunology/Inflammation related
Interleukin
IRAK
LTR
MALT
MRP
MyD88
NADPH-oxidase
NO Synthase
NOD
NOD-like Receptor (NLR)
NOS
Nrf2
PGE Synthase
Prostaglandin Receptor
Reactive Oxygen Species
ROS
STAT
ERK
JNK
KLF
MAPK
MEK
MLK
MNK
p38 MAPK
Raf
Ras
S6 Kinase
TOPK
ABC
Anion Exchanger
Aquaporin
ATPase
BCRP
Calcium Channel
CFTR
Chloride channel
CRM1
GABA Receptor
HCN Channel
iGluR
Monoamine Transporter
Monocarboxylate transporter
Na+/Ca2+ Exchanger
Na-K-Cl cotransporter
NPC1L1
OAT
OCT
P2X Receptor
P-gp
Piezo Channel
Potassium Channel
Proton pump
Sodium Channel
TRP/TRPV Channel
VDAC
Acetyl-CoA Carboxylase
aconitase
Acyltransferase
Adenosine deaminase
Amino Acids and Derivatives
Aminopeptidase
Amylase
Angiotensin-converting Enzyme (ACE)
ATP Citrate Lyase
Carbonic Anhydrase
Casein Kinase
CETP
CPT
Decarboxylase
Dehydrogenase
Drug Metabolite
Endogenous Metabolite
Epoxide Hydrolase
FAAH
FABP
Factor Xa
Fatty Acid Synthase
FXR
Glucokinase
Glucosidase
Glutathione Peroxidase
Glycosidase
GSNOR
hCE
Hexokinase
HMG-CoA Reductase
Hydrogenase
Hydroxylase
IDO
Indoleamine 2,3-Dioxygenase (IDO)
Antibacterial
Antibiotic
Antifection
Antifungal
Anti-infection
GluCls
HBV
HCV Protease
HIV Protease
HSV
Influenza Virus
Integrase
Parasite
Reverse Transcriptase
Ribosome
RSV
SARS-CoV
Virus Protease
AAK1 (AP2 associated kinase 1)
AChE
AChR
Adenylyl cyclase
BACE
Beta Amyloid
Beta-Secretase
CaMK
Gamma-secretase
GluR
GlyT
Huntingtin
Imidazoline Receptor
MAO
Monoamine Oxidase
MT Receptor
Neuropeptide Y Receptor
NMDAR
NMU2R
Norepinephrine
OX Receptor
P2Y Receptor
Serotonin Transporter
IκB/IKK
NF-κB
RANKL/RANK
Antioxidant
CAT
Free radical scavengers
Glutathione reductase
GPX
GST
GSK-3
MELK
mTOR
PDK
PI3K
PI4K
PTEN
SIK
ATTECs
AUTACs
E3 连接酶配体 - Linker 偶联物
E3 连接酶配体
Ligands for Target Protein for PROTAC
LYTACs
Molecular Glues
PROTAC Linker
PROTACs
SNIPERs
靶蛋白配体-linker 偶联物
Carboxypeptidase
Cysteine Protease
DPP-4
Glutaminase
MMP
Prolyl Endopeptidase (PREP)
Protease
Proteasome
Serine Protease
Thrombin
Tyrosinase
Hippo pathway
Porcupine
Smo
Stemness kinase
TGF-beta/Smad
YAP
ACK
c-Fms
c-Kit
c-Met/HGFR
CSF-1R
Discoidin Domain Receptor (DDR)
Ephrin Receptor
Hck
IGF-1R
PKA
PYK2
ROS Kinase
TAM Receptor
Tie-2
Trk receptor
Tyrosine Kinases
E1/E2/E3 Enzyme
p97
PROTAC-Linker Conjugates for PAC
Drug-Linker Conjugates for ADC
Antibody-Drug Conjugates (ADCs)
ADC Linker
ADC Cytotoxin
ADC Antibody
热门靶点推荐
化合物库推荐
虚拟筛选
分子对接
分子动力学模拟
药物设计和结构优化
超高通量虚拟筛选算法
微秒级长时模拟超算平台
新型蛋白质/多肽药物设计
结合亲和力数据库-PDBbind
小分子数据库-TOPSCIENCE DATABASE
分子砌块
基于靶点筛选
基于表型筛选
药代动力学研究
DNA编码化合物库筛选
人工智能靶点鉴定
化学蛋白质组学靶点鉴定
网络药理学
版权所有©2015-2025 TargetMol Chemicals Inc.保留所有权利.
沪ICP备20019793号-4沪公网安备 31010602006700号




 您的购物车当前为空
您的购物车当前为空







 嗨!很高兴为您提供帮助!
嗨!很高兴为您提供帮助!