

购物车
- 全部删除
 您的购物车当前为空
您的购物车当前为空

 很棒
很棒SAR-020106 是一种强效、ATP 竞争性和选择性 CHK1 抑制剂,IC50为 13.3 nM。它对 CHK2 具有良好的选择性,可通过选择抗癌药物增强抗肿瘤活性。

为众多的药物研发团队赋能,
让新药发现更简单!
很棒SAR-020106 是一种强效、ATP 竞争性和选择性 CHK1 抑制剂,IC50为 13.3 nM。它对 CHK2 具有良好的选择性,可通过选择抗癌药物增强抗肿瘤活性。
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1 mg | ¥ 413 | 现货 | |
| 5 mg | ¥ 980 | 现货 | |
| 10 mg | ¥ 1,570 | 现货 | |
| 25 mg | ¥ 3,380 | 现货 | |
| 50 mg | ¥ 4,820 | 现货 | |
| 100 mg | ¥ 6,690 | 现货 | |
| 1 mL x 10 mM (in DMSO) | ¥ 1,180 | 现货 |
| 产品描述 | SAR-020106 is a potent, ATP-competitive, and selective CHK1 inhibitor with an IC50 of 13.3 nmol/L on the isolated human enzyme. |
| 靶点活性 | Chk1:13.3 nM (IC50) |
| 体外活性 | SAR-020106 在裸鼠体内增强了 SW620 人结肠癌异种移植物对伊立替康和吉西他滨的疗效[2]。 |
| 体内活性 | SAR-020106是一种与ATP竞争的、强效且选择性的CHK1抑制剂,对分离的人类酶的IC(50)为13.3 nmol/L。该化合物能够以IC(50)为55 nmol/L的效力,抑制HT29细胞中由依托泊苷引发的G(2)阶段停滞,并在体外多种结肠肿瘤细胞线中,以p53依赖的方式显著增强吉西他滨和SN38的细胞杀伤作用,增强倍数为3.0至29倍。生物标志物研究表明,SAR-020106能够剂量依赖性地抑制细胞毒药物诱导的CHK1在S296上的自磷酸化,以及在体外和体内阻断CDK1在Y15上的磷酸化。细胞毒化合物组合与gammaH2AX增加和poly ADP核糖聚合酶切割增强相关,这与SAR-020106增强的DNA损伤和肿瘤细胞死亡一致。伊立替康和吉西他滨的抗肿瘤活动通过SAR-020106在体内得到增强,且毒性最小。SAR-020106代表了一种新型CHK1抑制剂,能够在体内增强选定抗癌化合物的抗肿瘤活性,因此可能具有临床应用价值[1]。 |
| 分子量 | 382.85 |
| 分子式 | C19H19ClN6O |
| CAS No. | 1184843-57-9 |
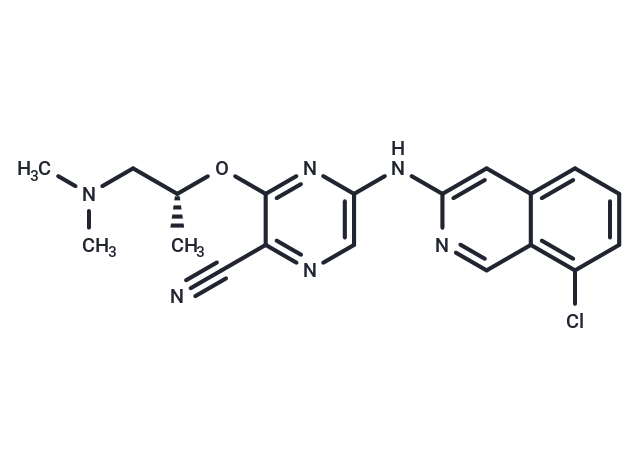
| Smiles | C[C@H](CN(C)C)Oc1nc(Nc2cc3cccc(Cl)c3cn2)cnc1C#N |
| 密度 | no data available |
| 存储 | Powder: -20°C for 3 years | In solvent: -80°C for 1 year | Shipping with blue ice. | |||||||||||||||||||||||||
| 溶解度信息 | DMSO: 12 mg/mL (31.34 mM) | |||||||||||||||||||||||||
溶液配制表 | ||||||||||||||||||||||||||
| ||||||||||||||||||||||||||
 嗨!很高兴为您提供帮助!
嗨!很高兴为您提供帮助! 比如您的给药剂量是 10 mg/kg ,每只动物体重 20 g ,给药体积 100 μL ,
比如您的给药剂量是 10 mg/kg ,每只动物体重 20 g ,给药体积 100 μL , 

评论内容